The basic premise for using thermofluor for drug screening assumes that high energy binding sites on proteins for small molecules (e.g. drugs) are functionally significant.
These include:
-Enzyme active sites
-Cofactor binding sites
-Allosteric effector binding sites
-Protein:Protein interaction sites that bind adaptor peptide domains (e.g. SH2, SH3, etc.)
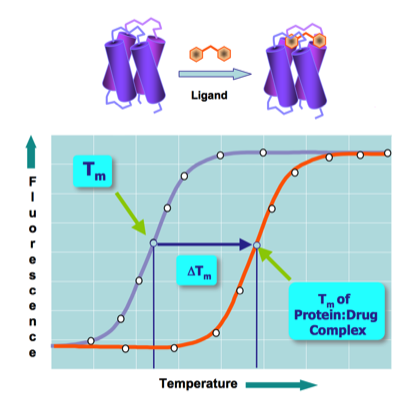
The free energy of ligand binding to the native state of a protein (assuming a substantial enthalpy contribution) produces an increase (positive Delta Tm) in the protein's thermal stability that can be measured using thermofluor. Thermoflour technology has consequently been used in numerous drug-screening campaigns to find compounds that range from enzyme inhibitors to agents that interfere with protein-protein interactions.
For ligand binding experiments, it is necessary to saturate the target protein binding site, so that solution ligand concentrations typically have to be 50-100um to find binding “hits” in the 5-10uM range. As a result of this solubility requirement, thermofluor HTS campaigns of “drug-like” screening libraries are typically carried out with relatively pure single compounds to insure adequate solubility under most solution assay conditions.
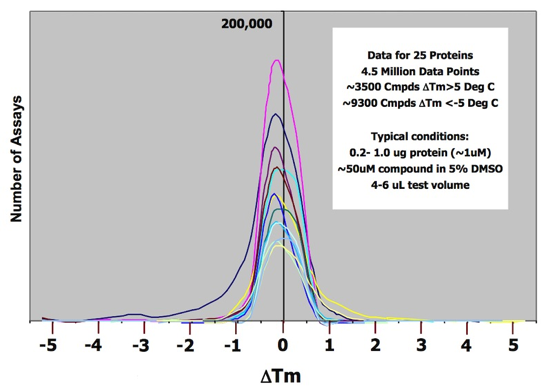
Most thermofluor screening campaigns produce “destabilizing” hits (e.g Delta Tm is negative) in addition to stabilizing hits (Delta Tm positive), presumably reflecting preferential ligand binding to partially unfolded protein states. Many such compounds may appear as active hits in corresponding enzymatic assays of target activity, but rarely develop extensive SAR relationships, and are often shown to act promiscuously against multiple protein targets.