Publications
D. Molecular Modeling & Simulation

Model Beta Coil
My work on recurring motifs in protein structures sparked an interest in understanding correlated motions and how these might be involved in protein recognition and function. Horizons expanded when I moved to DuPont Central Research, where in addition to a core crystallography group (Barry Finzel, Doug Ohlendorf, & Pat Weber), I was able to assemble an outstanding group of computationally (John Wendoloski & Zelda Wasserman) and graphically (Dick Hilmer) sophisticated collaborators. In addition we were able to recruit Mike Levitt as an advisor to the group.
The availability of a CRAY XMP and other supercomputing hardware at DuPont allowed us to carry out a number of pioneering MD investigations of biological systems, including electron transfer reactions, micelles, and biological elastomers.
The availability of a CRAY XMP and other supercomputing hardware at DuPont allowed us to carry out a number of pioneering MD investigations of biological systems, including electron transfer reactions, micelles, and biological elastomers.
26. Conformational Flexibility and Amide Exchange Stability in Protein ß-Sheets.
F.R. Salemme, Nature 1982; 299: 754-756
F.R. Salemme, Nature 1982; 299: 754-756
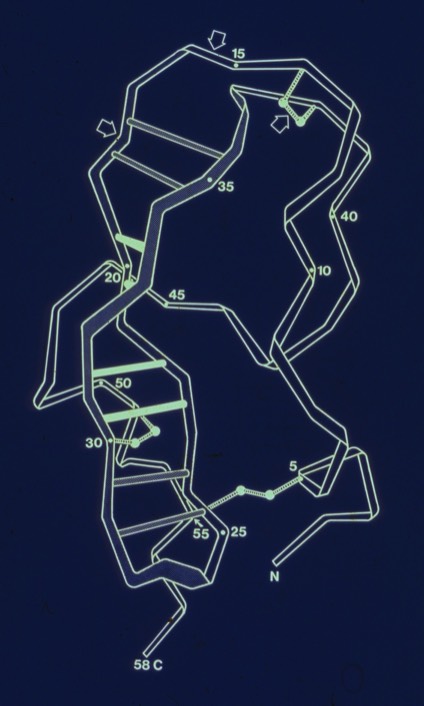
Fred Richards had shown that amide proton exchange rates generally correlated with solvent exposure in protein structures. Kurt Wuthrich's NMR studies of the small protein BPTI nevertheless showed that some H-bonds in the relatively exposed two-stranded antiparallel sheet were very stable to exchange. My paper discussed the possibility that owing to the structure of the antiparallel sheet, it could accommodate relatively large scale excursions from the ground state while keeping the H-bonds perfectly intact.
27. A Model for Catabolite Activator Protein Binding to Supercoiled DNA.
F.R. Salemme Proc. Natl. Acad. USA 1982; 79: 5263-5268
F.R. Salemme Proc. Natl. Acad. USA 1982; 79: 5263-5268
44. Computer Simulations of Biological Interactions and Reactivity.
J.J. Wendoloski, Z. Wasserman, F.R. Salemme, J. Computer Aided Molecular Design 1987; 1: 313-322.
J.J. Wendoloski, Z. Wasserman, F.R. Salemme, J. Computer Aided Molecular Design 1987; 1: 313-322.
45. Molecular Dynamics of a Cytochrome c-Cytochrome b5 Electron Transfer Complex.
J.J. Wendoloski, J.B. Matthew, P.C.Weber, and F.R. Salemme, Science 1987; 238: 794-796.
J.J. Wendoloski, J.B. Matthew, P.C.Weber, and F.R. Salemme, Science 1987; 238: 794-796.
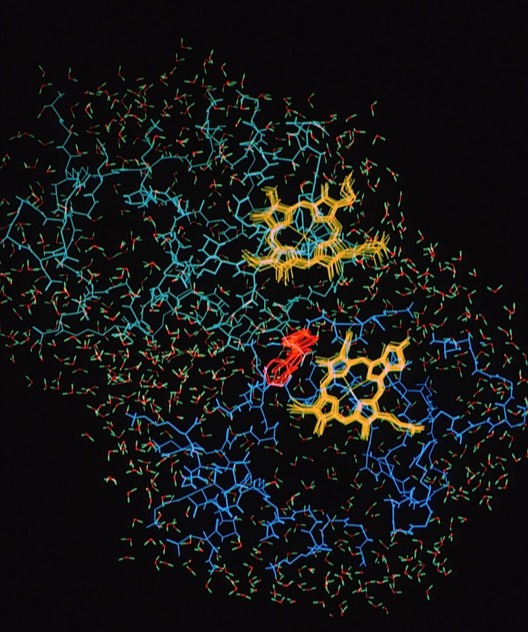
This study picked up on the static modeling study of the cytochrome c- cytochrome b5 electron transfer complex (Ref 9) and provided some important insights into the electron transfer process. Mainly these devolved around 1) the extent of intermolecular motions, 2) the effect of intermolecular electrostatic interactions in stabilizing the overall complex geometry, and 3) he motions of aromatic residue side chains that formed a putative inter-heme electron tunneling pathway.
The simulation was computed using the AMBER potential and MD code and included atomic models of both proteins and an extensive hydration shell of discrete water molecules, making it one of the larger scale computations at the time.
The simulation was computed using the AMBER potential and MD code and included atomic models of both proteins and an extensive hydration shell of discrete water molecules, making it one of the larger scale computations at the time.
49. Structural Polymorphism in Transmembrane Channels.
F.R. Salemme, Science 1988; 241: 145 & 190.
F.R. Salemme, Science 1988; 241: 145 & 190.
52. Molecular Dynamics Simulation of a Phospholipid Micelle, J.J. Wendoloski, S. Kimatian, C.E. Schutt, F.R. Salemme, Science 1989; 243: 636-638.
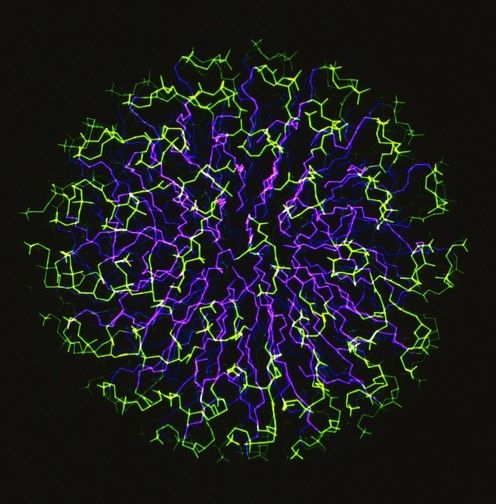
During my DuPont years, I had the opportunity to work with my friend Clarance Schutt at Princeton, who among other things, interned several talented Princeton undergrads over the summers in my lab. As one project, we undertook a simulation of a completely hydrated phospholipid micelle. We observed somewhat unexpected results in the simulation, which looked a bit like plate tectonics with locally ordered phospholipid rafts, separated by regions of discontinuous packing interactions, shifting, forming and recombining throughout the simulation. Nevertheless, time averaging the dynamic behavior accurately reproduced NMR order parameter measurements, although these had been given a completely different physical interpretation (local spinning top model) in earlier work.
57. Modeling with Substructure Libraries Derived from Known Protein Structures, Finzel, BC, Kimatian S, Ohlendorf, DH, Wendoloski, JJ, Levitt M, Salemme FR, In Crystallographic and Modeling Methods in Molecular Design (S Ealick & C Bugg eds.) Springer Verlag, New York, 175-189 (1990)
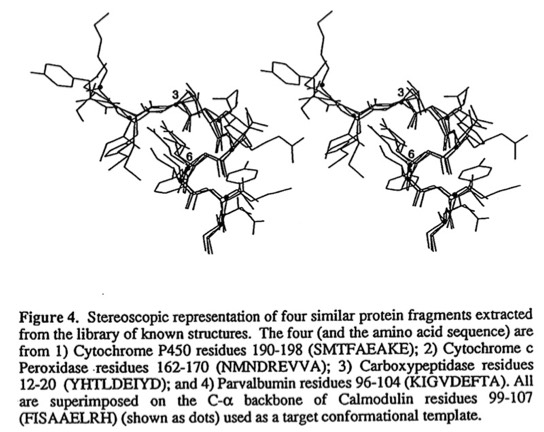
Barry Finzel developed the FRAGLE protein substructure modeling system at DuPont, which I believe was the first computer implementation of this now common feature of protein modeling programs. In my view, Barry's implementation is still among the most flexible and useful since 1) it found candidate substructures (both continuous and non continuous) by fitting overlapping pairs of Ca atoms at both ends of each substructure (in contrast, just fitting the end points in applications such a loop substitutions can result in physically unreasonable backbone conformations in the linking region) and 2) could be applied according to a very flexible set of superimposed amino acid sequence constraints that often revealed canonical sequence-structure motifs.
59 A Molecular Dynamics Investigation of the Elastomeric Restoring Force in Elastin, Z.W. Wasserman and F.R. Salemme, Biopolymers 1990; 29: 1613-1631.

DuPont had developed a substantial business around the elastomeric copolymer Lycra, initially used in bathing suits, but now widely used in many stretch fabric applications. This motivated studies in all-natural elastomeric polymers, which lead in turn to Zelda Wasserman's pioneering modeling study of elastin. Zelda's forced-displacement MD simulation clearly demonstrated how both changes in fiber hydration and conformational entropy contributed to fiber elastomeric properties when it was stretched.
60. Competitor Analogs for Defined T Cell Antigens: Peptides Incorporating a Putative Binding Motif and Polyproline or Polyglycine Spacers
J.L. Maryanski, A.s. Verdini, P.C. Weber, F.R. Salemme, G. Corradin Cell 1990 60:63-72
J.L. Maryanski, A.s. Verdini, P.C. Weber, F.R. Salemme, G. Corradin Cell 1990 60:63-72
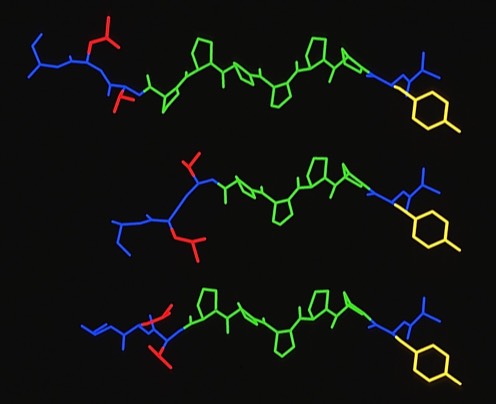
This was one of two modeling studies of T-Cell antigens that arose from collaborations with Giampietro Corradin at the University of Lausanne (See also 66). The availability of the MHC structure from Don Wiley's lab at Harvard was a key catalyst for such studies which were ultimately aimed at finding compounds that would potentially modulate MHC-mediated autoimmune responses.
62. Computational and database retrieval approaches for determining polypeptide conformation.
F. R. Salemme, Z. Wasserman, and P. C. Weber Proteins Structure, Dynamics and Design (V. Renugopalakrishnan, P. Carey, I. Smith, S. Huang, A. Storer, Eds.) ESCOM Science Publishers, Leiden 1991; 260-265.
F. R. Salemme, Z. Wasserman, and P. C. Weber Proteins Structure, Dynamics and Design (V. Renugopalakrishnan, P. Carey, I. Smith, S. Huang, A. Storer, Eds.) ESCOM Science Publishers, Leiden 1991; 260-265.
65. Cooperative Ligand Reorientations in Cytochrome c3: A Molecular Dynamics Simulation.
P.C. Weber, J.J. Wendoloski and F. R. Salemme Biochim. Biophys. Acta 1991; 1058: 83-84.
P.C. Weber, J.J. Wendoloski and F. R. Salemme Biochim. Biophys. Acta 1991; 1058: 83-84.
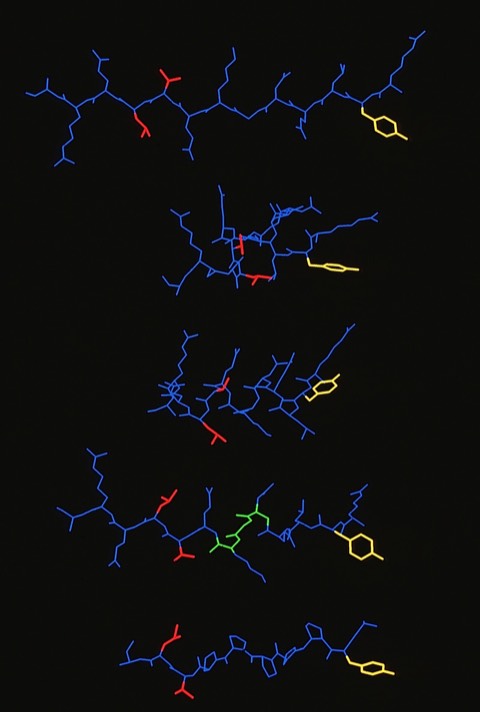
66. The Identification of Tyrosine as a Common Key Residue in Unrelated H-2Kd Restricted Antigenic Peptides.
J. Maryanski, P. Romero, A. Van Pel, , T. Boon, F.R. Salemme, J-C Cerrotitini, G. Corradin, International Immunology 1991; 3: 1035-1042.
J. Maryanski, P. Romero, A. Van Pel, , T. Boon, F.R. Salemme, J-C Cerrotitini, G. Corradin, International Immunology 1991; 3: 1035-1042.
This modeling study examined how evidently sequentially unrelated peptides could conformationally present the same feature to an MHC binding site.
73. PROBIT: A Statistical Approach to Modeling Proteins from Partial Coordinate Data Using Substructure Libraries.
J.J. Wendoloski & F.R. Salemme J. Molecular Graphics 1992; 10: 124-126.
J.J. Wendoloski & F.R. Salemme J. Molecular Graphics 1992; 10: 124-126.
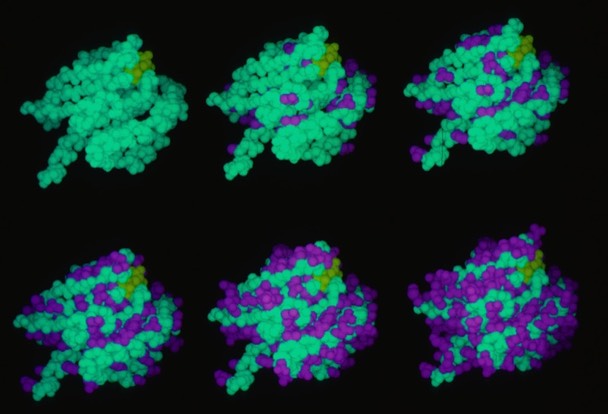
PROBIT was an automated protein modeling program developed at DuPont that was motivated by the observation that amino acid side-chains had well defined side chain rotomer distributions. As the number of high resolution protein X-ray structures began to proliferate, it became possible to compile rotomer statistics of side chains in secondary structure context (e. g. branched aliphatic side chains can only occupy one chi-one rotomer state when in an alpha helix). PROBIT rebuilt side chains in order of statistical rotomer probability and then effectively used the "prepacked" residiues to find good packing arrangements for more "ambiguous" residues (which often turned out to be aromatic side chains). Again, features of PROBIT turn up in many current protein modeling systems.