Modeling & Simulation
MOLECULAR MODELS
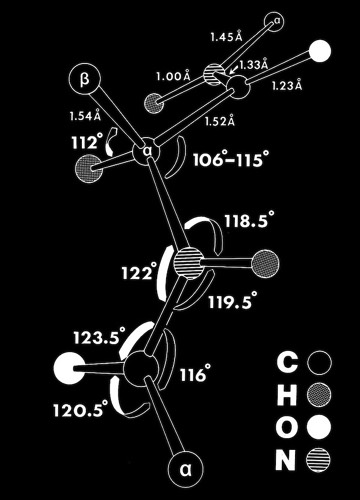
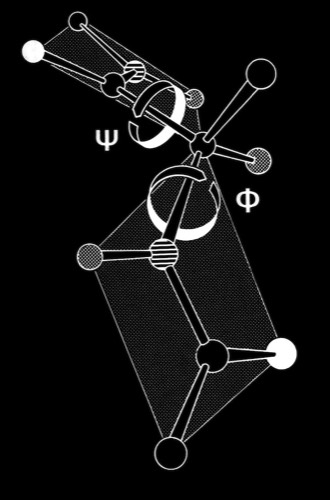

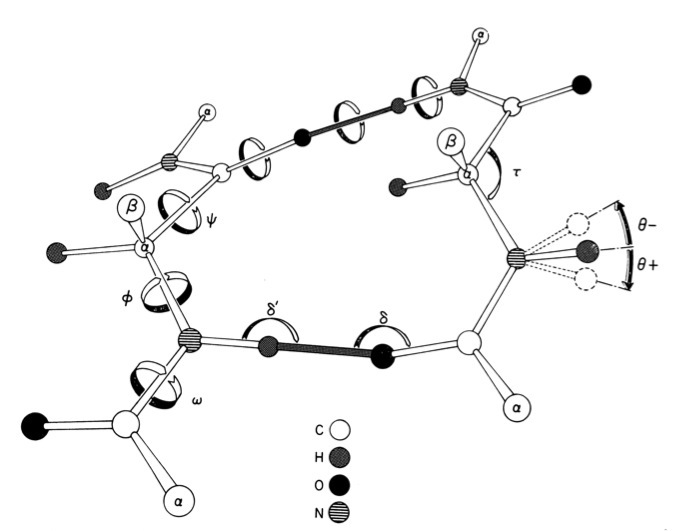
In the days before computer graphics, most studies of protein structure used physical models (which have a long tradition in structural biology), most famously the 1cm/Angstrom Kendrew models used to build protein structural models “into” electron density maps using a Richards Box.
I built some very large-scale (10cm/Angstrom) polypeptide models with many additional conformational degrees of freedom to study beta sheets. These were instrumental in developing computer methods that allowed Dave Weatherford and me to produce computer models of various beta sheet conformations from first mechanical principles that very closely approximated the great variety if twisted sheet conformations observed in protein structures.
At Arizona, I was one of the early recipients of the NIH-sponsored MMSX computer graphics system, originally developed at the Washington University Medical School. This system made the generation of hypothetical electron transfer complexes such as the cytochrome c - flavodoxin complex that we studied in collaboration with Gordon Tollin and Jim Matthews relatively straightforward, compared to earlier approaches.
I built some very large-scale (10cm/Angstrom) polypeptide models with many additional conformational degrees of freedom to study beta sheets. These were instrumental in developing computer methods that allowed Dave Weatherford and me to produce computer models of various beta sheet conformations from first mechanical principles that very closely approximated the great variety if twisted sheet conformations observed in protein structures.
At Arizona, I was one of the early recipients of the NIH-sponsored MMSX computer graphics system, originally developed at the Washington University Medical School. This system made the generation of hypothetical electron transfer complexes such as the cytochrome c - flavodoxin complex that we studied in collaboration with Gordon Tollin and Jim Matthews relatively straightforward, compared to earlier approaches.
STRUCTURAL CONSERVATION DURING PROTEIN EVOLUTION
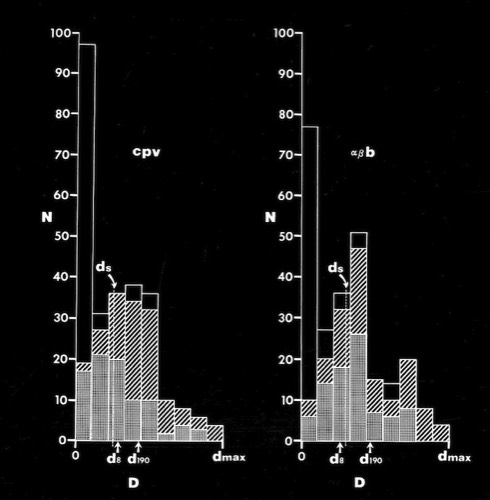
In the early days of protein structure and sequence determination, it was unclear whether 40% amino acid sequence homology suggested that two protein structures would be structurally similar or completely different (Of course, now we recognize that sequence homology of 20% or less, depending on context, can reflect structural and functional homology). One idea with currency in the early days of structural biology was that the genetic code for the amino acids tended to insure that single base codon mutations minimized possible disruptions to protein structure and function. At Arizona, I looked at this problem with one of my first graduate students, Steve Jordan, and a talented undergraduate, Mike Miller.
We characterized the amino acids in terms of relevant structural properties and then characterized the consequent changes in structural properties that occurred with single base mutations. In fact we found that although some single-base changes were conservative, many others were not, and that the fitness landscape of single nucleotide codon substitutions was generally discontinuous.
We characterized the amino acids in terms of relevant structural properties and then characterized the consequent changes in structural properties that occurred with single base mutations. In fact we found that although some single-base changes were conservative, many others were not, and that the fitness landscape of single nucleotide codon substitutions was generally discontinuous.
ELECTROSTATIC INTERACTIONS
My interactions with Jim Matthew while on sabbatical at Yale subsequently lead to our working together in the Protein Engineering Division at Genex Corporation, and later at DuPont Central Research. We applied a computational method for computing and displaying electrostatic fields that had been developed by Jim to a variety of biochemical systems. A particularly instructive representation was the “2kT potential surface” which represented the surface on which a randomly moving countercharge would be effectively bound in the vicinity of the field. This provided an indication of how close two proteins had to approach each other to be influenced by each other'e surface charge.
Electron Transfer Reactions
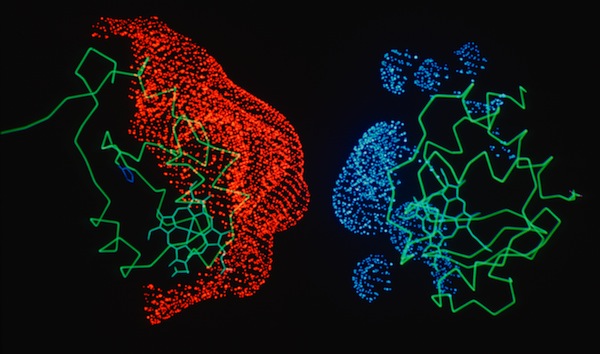
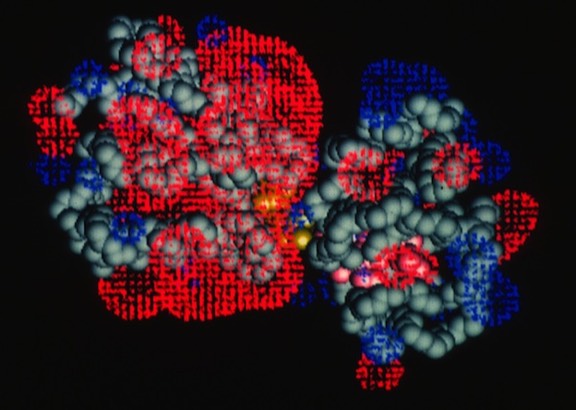
Work on the role of electrostatics on protein-protein interactions was initially motivated by structural observations for several redox proteins systems that were known to undergo electron transfer reaction that could be attenuated by increases in solution ionic strength. In addition to redox proteins (described here), we usefully applied electrostatic field calculations to systems like calmodulin and protein DNA interactions.
Studies on electrostatically facilitated ET interactions included computational and modeling studies of the Cytochrome C-Cytochrome b5 and Cytochrome C- flavodoxin complexes.
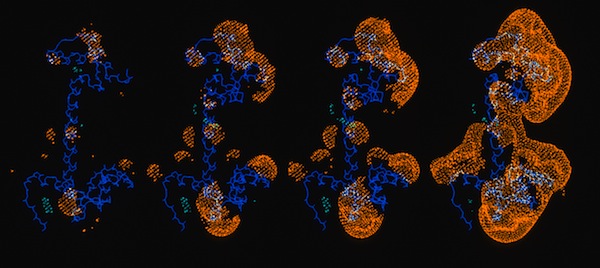
Calmodulin
PROTEIN-DNA INTERACTIONS
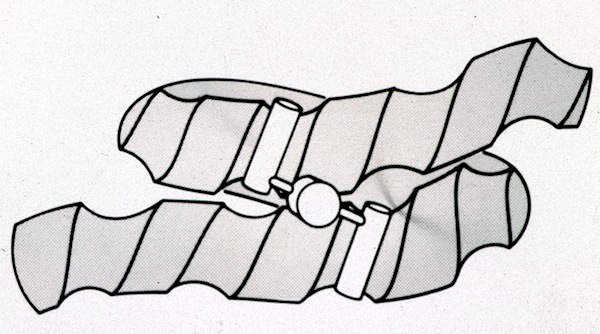
While on Sabbatical at Yale in 1981, I worked on several projects, mostly of a theoretical nature. At the time the “helix into DNA groove” models of protein-DNA interaction were causing a lot of excitement in the structural biophysics world. Tom Steitz had just completed the structure of catabolite activator protein (CAP), a dimer with two alpha helices evidently oriented to fit the grooves of a left-handed DNA conformation that had earlier been described by Alex Rich.
I modeled a conjectural structure that envisioned the CAP dimer binding to supercoiled DNA, reasoning that grooves on adjacent coils of right-handed DNA would fit the helices in a dimer with helices apparently oriented to straight helices of left-handed DNA. A later CAP-DNA X-ray structure showed a complex where the DNA was sharply bent around the protein dimer.
I modeled a conjectural structure that envisioned the CAP dimer binding to supercoiled DNA, reasoning that grooves on adjacent coils of right-handed DNA would fit the helices in a dimer with helices apparently oriented to straight helices of left-handed DNA. A later CAP-DNA X-ray structure showed a complex where the DNA was sharply bent around the protein dimer.
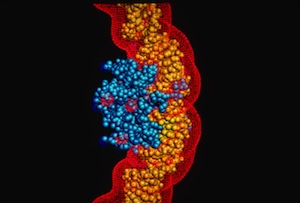

Work on protein-DNA interactions at Genex was lead by Doug Ohlendorf. Doug had determined the structure of the CRO protein when working in Brian Matthews lab at the University of Oregon prior to joining the Protein Engineering Group at Genex, and later DuPont Central Research.
Doug also modeled the streptavidin-DNA complex used to illustrate our article prepared for CRAY Channels.
Doug also modeled the streptavidin-DNA complex used to illustrate our article prepared for CRAY Channels.

CRYSTAL LATTICES
At DuPont Central Research, I was able to assemble an outstanding group of scientists using both experimental X-ray methods and computational simulations to investigate the structural properties of materials ranging from synthetic polymers to proteins. The DuPont environment was unusual in that we had extraordinary access to state-of-the-art computing resources including CRAY supercomputers, a substantial DEC VAX system, and a STAR ST100 Array Processor. An important component of our research effort was the development of novel capabilities using computer graphics. A key architect for several of these developments was Richard Hilmer, who was an expert in computer graphics programming using the Evans and Sutherland function network protocols. His achievements included the development of the MOLEDITOR program allowing the flexible display of protein molecular dynamics, including useful features such as the dynamic representation of hydrogen and ionic bond formation. Later, Dick expanded MOLEDITOR to incorporate a lattice modeling system that provided full six-degree of freedom control of molecular substructures in lattices with any plane or 3D crystallographic symmetry and/or desired cell dimensions. In addition, the system allowed the generation, display, and manipulation of two independent lattices simultaneously, a feature that was used to model and examine interactions in polymer or crystal lattices with interstitial host molecules. He also created the capability to generate images of crystal habits and faces superimposed on lattices of molecular structures. His codes were used to create and illustrate many of the MD and crystal packing diagrams shown on this website.
Cytochrome C-prime Bond Chains
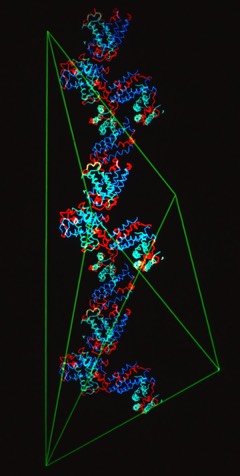
The structural refinement of cytochrome c-prime at 1.67 Angstrom resolution raised some interesting questions. Although the protein was a dimer of identical chains related by a dyad symmetry axis, the protein packed with a dimer in the asymmetric unit of the orthorhombic P212121 cell. Analysis of the comparative B-values between the monomers showed that the B-value differences between monomers could be explained as a consequence of static or dynamic rotational disordering of a chain of dimers related by 21 symmetry aligned parallel to a unit cell edge.
Lysozyme Bond Chains
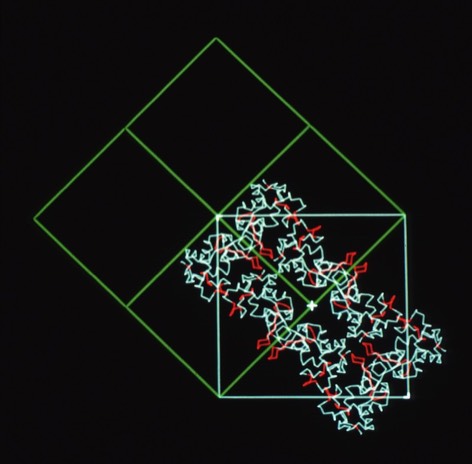

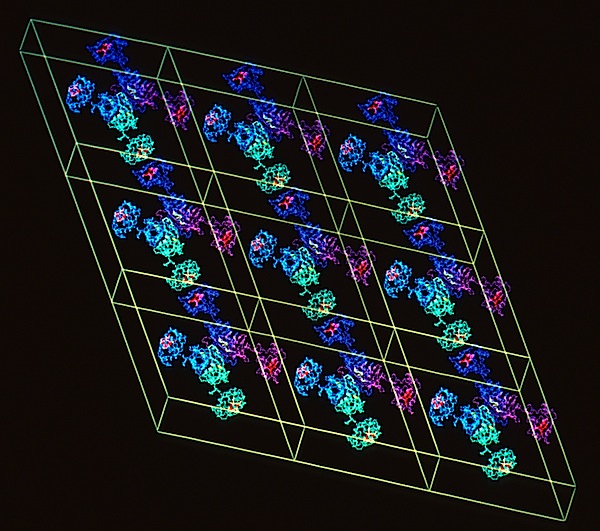
I subsequently became interested in the relationship between crystal habit and molecular interactions in protein crystals. To investigate this, I suggested to Lars Geneiser (at that time a Princeton undergraduate working at DuPont as a summer intern), that we might look at molecular interactions in a protein system where the protein crystallized in multiple space groups. At that point, the hen egg white lysozyme structure had been determined in four space groups: tetragonal (P43212), monoclinic (P21), triclinic (P1), and orthorhombic (P212121). Using the graphics tool developed by Dick Hilmer at CRD, Lars was able to construct the relevant crystal packing schemes and observed that similar molecular chains recurred in the alternative lysozyme crystal forms.
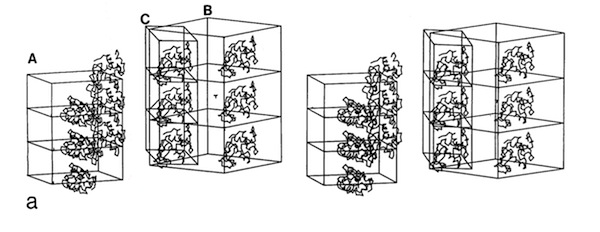
Recurrent Lysozyme Bond Chains
Crystal Lattices & Habits
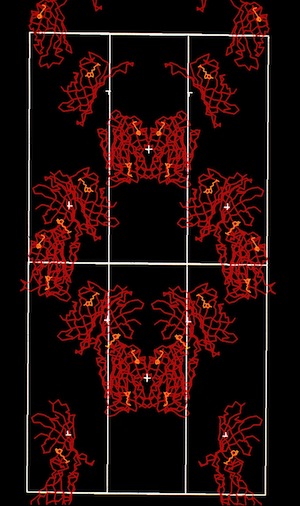
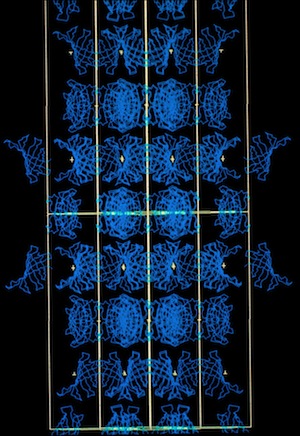
Synthetic Fibers & A Molecular Conductor

At DuPont Central Research we investigated several potential applications of biotechnology for the development of new materials based on structural polymers found in nature. DuPont had developed and successfully commercialized such polymers as Nylon, Kevlar, and Lycra, creating an interest in biopolymers with high strength (spider silk) and elasticity (elastin). One project was undertaken to express repetitive silk sequences in E. coli. Although fibers were successfully spun from the high MW recombinant silk proteins, determination of the best spinning conditions that would produce fibers with natural silk-like tensile strength proved to be a challenging technical issue.
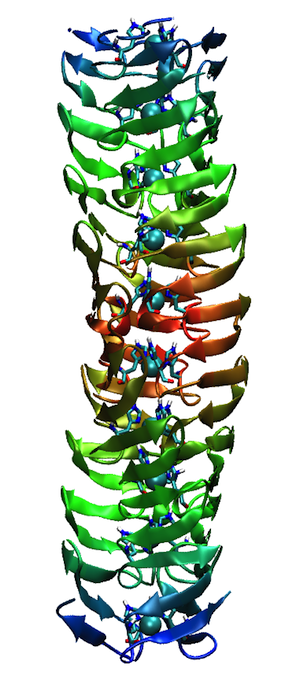
In a related effort, recombinant DNA technology was used to produce a stiff fibrous structure, composed of polypeptide chains organized as hairpin bends and inspired by the repetitive sequences observed in the trimeric adenovirus spike protein.
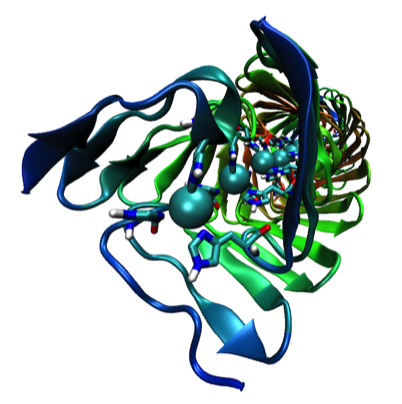
The adenovirus fiber-inspired modeling was carried out by Zelda Wasserman while working in my group. Zelda also extended the original design to model a molecular wire composed of 3 polypeptide chains incorporating repetitive hairpin bend sequences. In this design, each polypeptide chain also incorporated repetitive histidine residues whose imidazole side chains could coordinate to copper atoms forming the conductor core.
In a related effort, recombinant DNA technology was used to produce a stiff fibrous structure, composed of polypeptide chains organized as hairpin bends and inspired by the repetitive sequences observed in the trimeric adenovirus spike protein.
The adenovirus fiber-inspired modeling was carried out by Zelda Wasserman while working in my group. Zelda also extended the original design to model a molecular wire composed of 3 polypeptide chains incorporating repetitive hairpin bend sequences. In this design, each polypeptide chain also incorporated repetitive histidine residues whose imidazole side chains could coordinate to copper atoms forming the conductor core.
Lattice Modeling: Biomolecular Switch
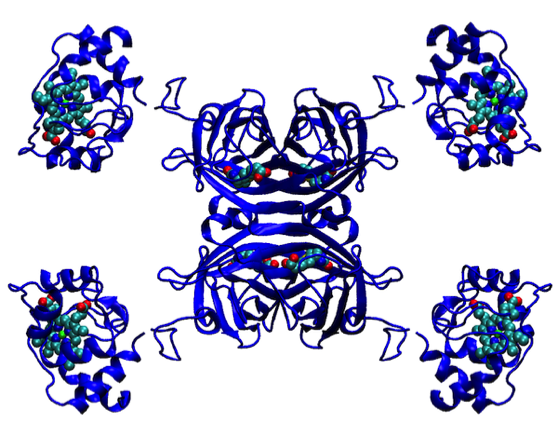
The flexible lattice building capabilities of Dick Hilmer’s Evans & Sutherland code allowed the development of concept studies for new functional materials based on protein engineering. One example shown here envisions a fusion of a c-type cytochrome with the polypeptide chains of the D2-symmetric streptavidin tetramer. We experimented with lattice constructions that could allow alternative chains of heme-heme interactions to be formed along alternative directions in the molecular lattice.

MOLECULAR DYNAMICS SIMULATIONS
The outstanding computational resources and talented collaborators that I had at DuPont CRD allowed our group to investigate many interesting phenomena using MD methods. We were also fortunate to have Michael Levitt, one of the originators of protein MD methods, as a consultant to our group.
Electron Transfer Proteins
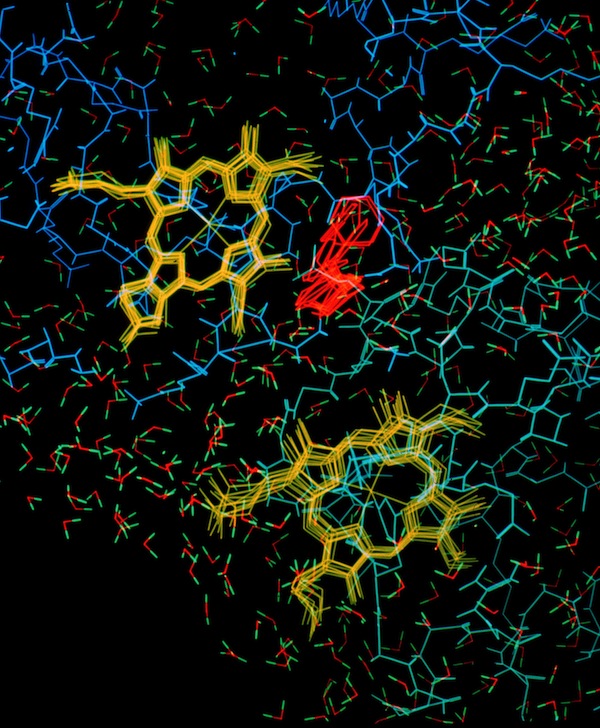
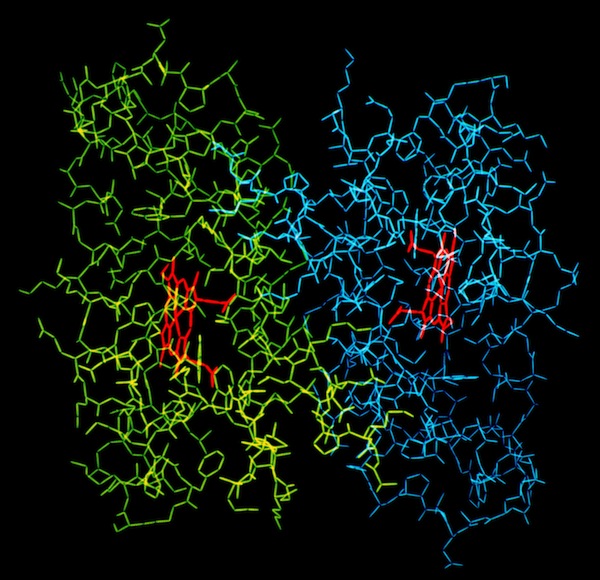
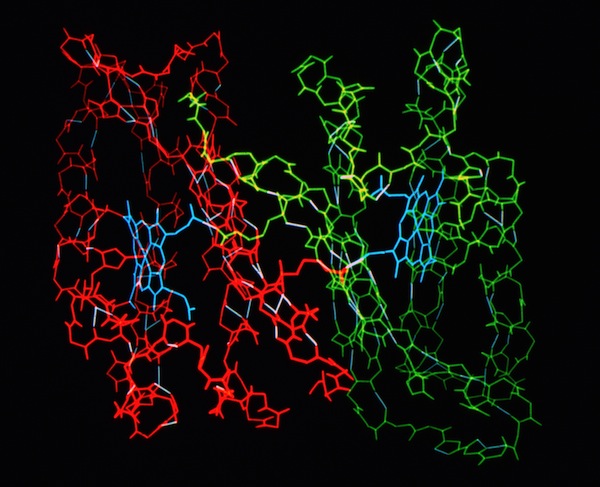
A long interest in electron transfer mechanisms motivated several simulations of electron transfer proteins, with particular relevance to redox complex ET transition states, conformational flexibility, and heme protein potential regulation. Examples included MD simulations of the cytochrome c-b5 complex, low-potential cytochromes c, and cytochrome c-prime.
Cytochrome C-prime Asymmetry
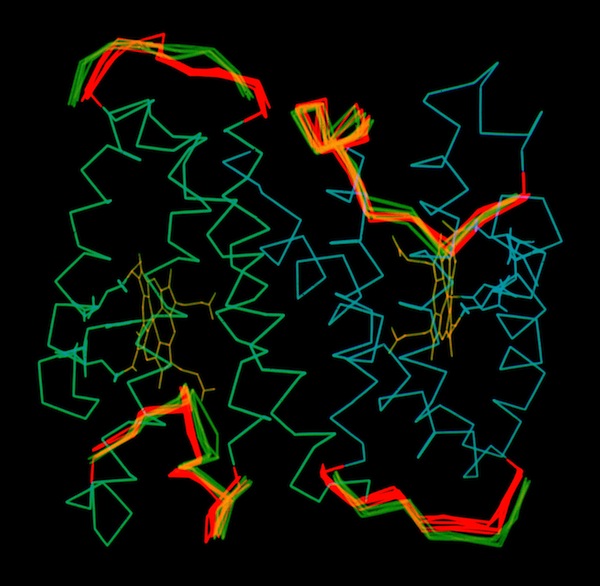
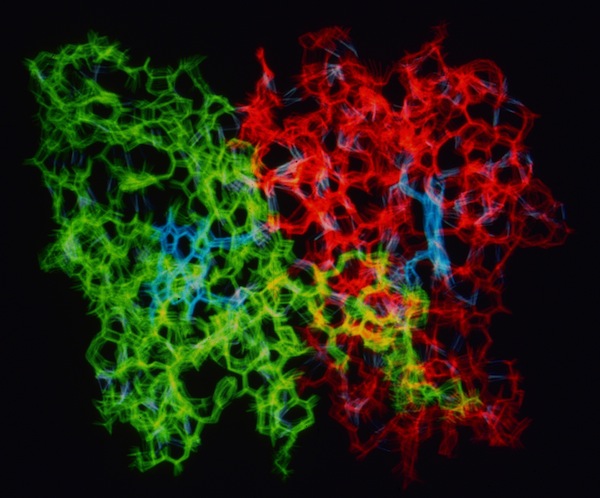
The structure determination of cytochrome c-prime demonstrated the presence of a dimer in the crystal asymmetric unit. This means that otherwise identical protein monomers make different interactions in forming the 3D crystal lattice. This could occur if local lattice interactions trapped different conformational states at the lattice interaction sites. To examine this possibility, and generally look at the possibility of correlated motions in s symmetric protein molecule, John Wendoloski carried out a hydrated MD simulation of the cytochrome c-prime dimer and Larry Greller analyzed the trajectory. Little evidence of correlated motion was found in the structure in the 300ps MD trajectory. Instead the system manifest behavior characteristic of many nonlinear dynamical systems. Nevertheless, it was observed that the surface loops where uncorrelated dynamic excursions were most different were also the sites of intermolecular lattice contacts.
The structure determination of cytochrome c-prime demonstrated the presence of a dimer in the crystal asymmetric unit. This means that otherwise identical protein monomers make different interactions in forming the 3D crystal lattice. This could occur if local lattice interactions trapped different conformational states at the lattice interaction sites. To examine this possibility, and generally look at the possibility of correlated motions in s symmetric protein molecule, John Wendoloski carried out a hydrated MD simulation of the cytochrome c-prime dimer and Larry Greller analyzed the trajectory. Little evidence of correlated motion was found in the structure in the 300ps MD trajectory. Instead the system manifest behavior characteristic of many nonlinear dynamical systems. Nevertheless, it was observed that the surface loops where uncorrelated dynamic excursions were most different were also the sites of intermolecular lattice contacts.
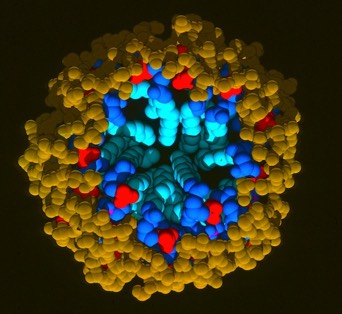
Phospholipid Micelle
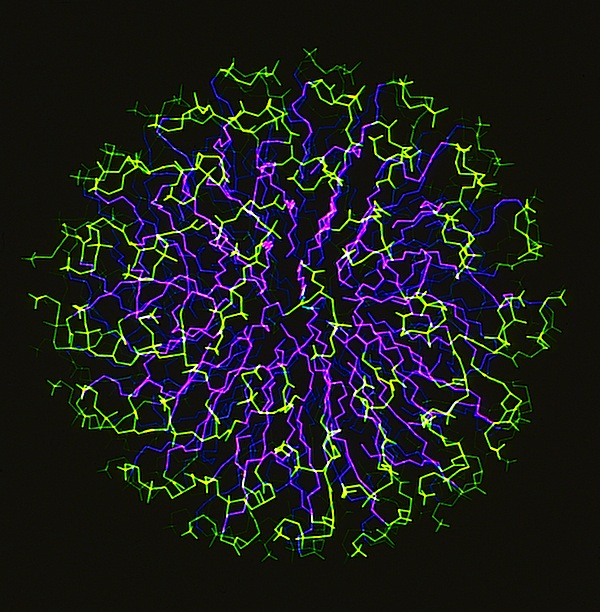
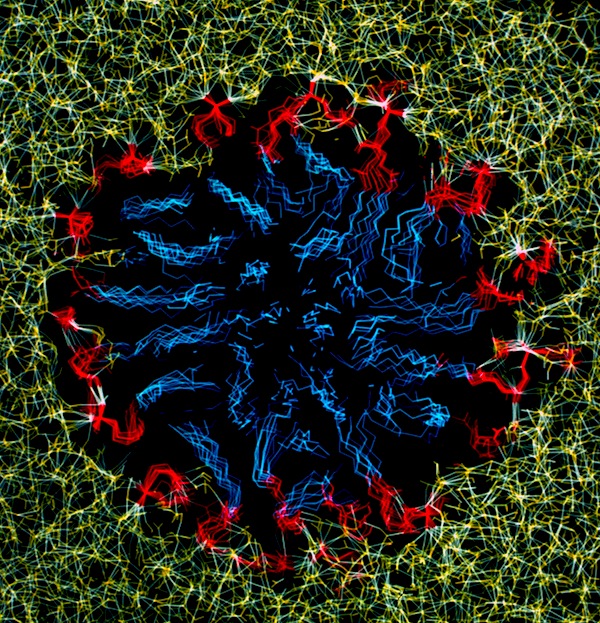
The phospholipid micelle simulation was a collaboration between members of our DuPont group (John Wendoloski and me), Clarence Schutt at Princeton, and a Princeton undergraduate summer intern, Steve Kimatian. The simulation showed somewhat surprising behavior. The then current micelle models assumed phospholipid motions that were relatively uniform where each phospholipid molecule described a time-averaged volume approximating a conical top (thus being consistent with close packing a spherical volume). In fact, the simulation showed the transient formation and dissolution of small phospholipid rafts separated by packing defects on the initially tightly packed micelle surface. Order parameters calculated from the MD trajectory nevertheless correlated well with experiment measurements made from micelle NMR studies. This represented one of those frequent cases in biological structure where the simple intuitive model is probably wrong.
Phospholipid Membranes
In addition to micelle simulations, we undertook simulations of phospholipid bilayers. These studies were viewed as first steps towards understanding membrane protein interactions.
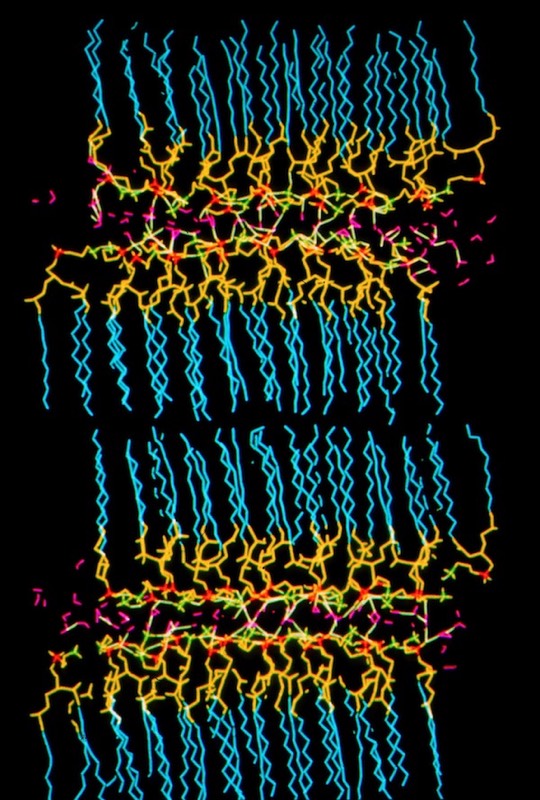
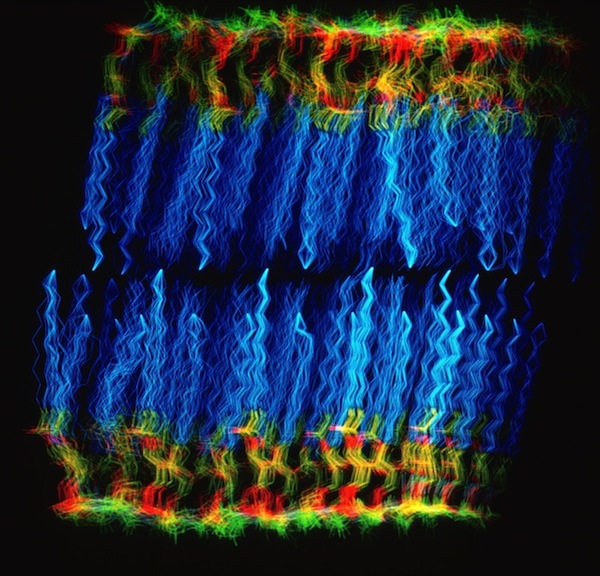

Phospholipase A2-Micelle Simulation
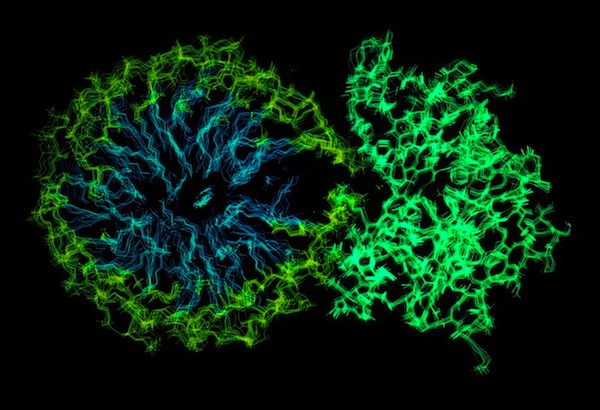
One of our early structure-based drug design programs at DuPont CRD was focused on finding inhibitors of phospholipase A2, an effort that lead us to carry out numerous X-ray crystal structures. PLA2 is an unusual enzyme in that it catalyzes cleavage of a substrate that is present in a micellar state. To gain insight into this phenomenon we performed an MD simulation of a hydrated phospholipid-PLA2 complex. Interestingly, the charged groups on the surface of PLA2 had the effect of disrupting the phospholipid organization, making the phospholipid scissile bond more accessible to the enzyme active site residues.
POLYMER MODELING & SIMULATION
Elastin
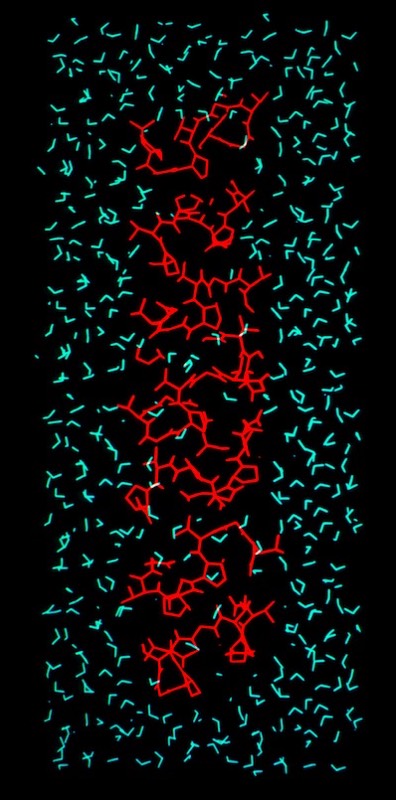
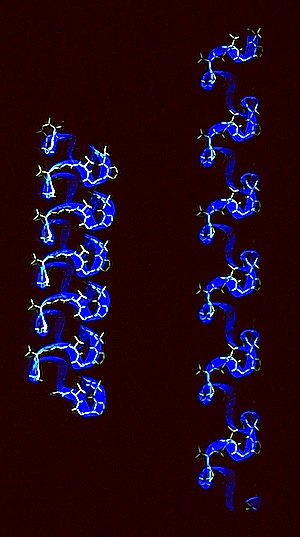
Given DuPont’s interest in elastomeric polymers like Lycra, and polymer physics in general, we were motivated use modeling and simulation methods to evaluate classical concepts of elastomeric restoring forces. Zelda Wasserman approached this problem by creating a molecular model for elastin, a natural elastomeric polymer that incorporates the repetitive pentapeptide sequence (VPGGVG)n, which was (and is) believed to form a continuous coil of stacked beta turns.
Zelda carried out a hydrated molecular dynamics simulation of the elastin coil as a function of stretch and used a treatment originated by Meirovich to evaluate the entropy changes occurring as the structure elongated. The results suggested that changing hydrophobic interactions contribute to elastin entropy as stretching is initiated, but that librational mechanisms assume greater importance in more extensively stretched conformations.
Zelda carried out a hydrated molecular dynamics simulation of the elastin coil as a function of stretch and used a treatment originated by Meirovich to evaluate the entropy changes occurring as the structure elongated. The results suggested that changing hydrophobic interactions contribute to elastin entropy as stretching is initiated, but that librational mechanisms assume greater importance in more extensively stretched conformations.
Kapton Lattice Modeling & Molecular Dynamics
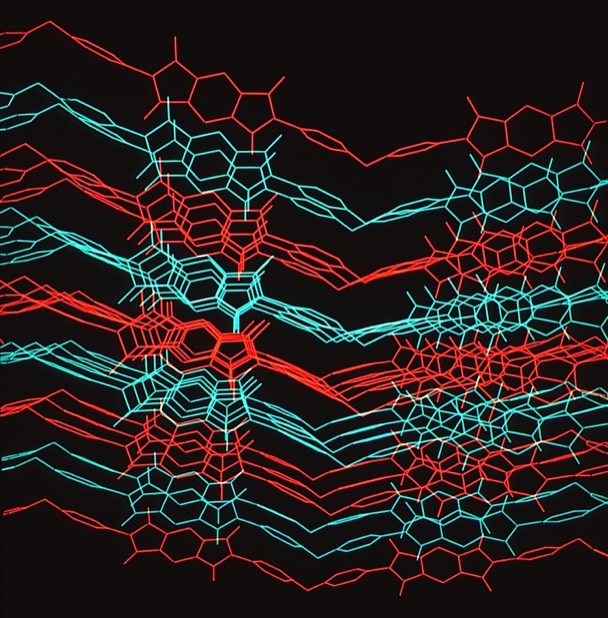
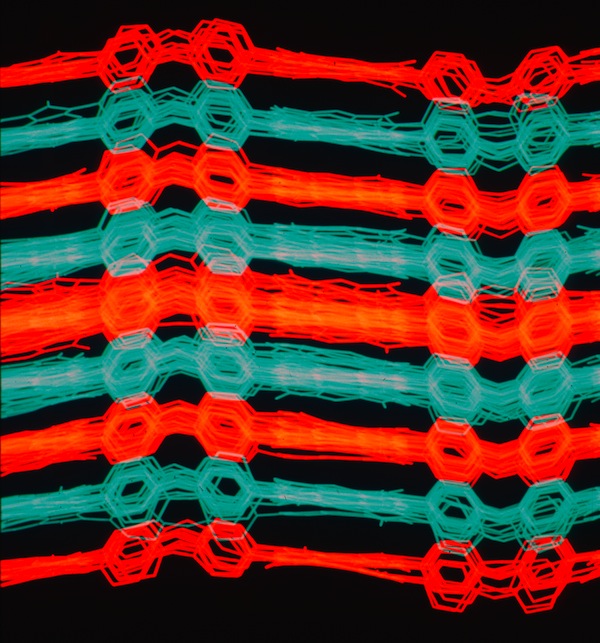
At Dupont Central Research we performed numerous modeling and MD studies of advanced polymers like Nylon, Kevlar, Nomex, and others that had been developed by DuPont scientists over the years. Model development was often guided by X-ray fiber diffraction studies. Many polymer modeling studies used the dual-lattice modeling capabilities developed by Dick Hilmer to model polymer co-solvent or polymer plasticizer interactions to better understanding polymer chain alignment during spinning processes and related polymer properties. One system we studied was Kapton, an advanced polyimide polymer used for high performance electronics and other applications. We used modeling tools to develop chain packing models and performed MD simulations that illuminated aspects of its high thermal stability.
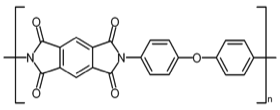
Kapton Polymer Unit