Structure-Based Drug Design
The use of 3D protein structural data to assist in drug design was an activity that closely followed the development of protein crystallography. For example, Max Perutz, who was among the first protein crystallographers, was interested in finding compounds that would modulate the oxygen binding characteristics of sickle-cell hemoglobin.
In many drug discovery programs, compounds that modulate protein activity are selected using a high-throughput assay and the binding properties of resulting hits optimized to preclinical leads through systematic chemical modification. The modification process is guided by computational tools that construct a structure-activity relationship (SAR) that attempts to infer the characteristics of the protein target binding site from the comparative properties of multiple lead compounds.
In many drug discovery programs, compounds that modulate protein activity are selected using a high-throughput assay and the binding properties of resulting hits optimized to preclinical leads through systematic chemical modification. The modification process is guided by computational tools that construct a structure-activity relationship (SAR) that attempts to infer the characteristics of the protein target binding site from the comparative properties of multiple lead compounds.
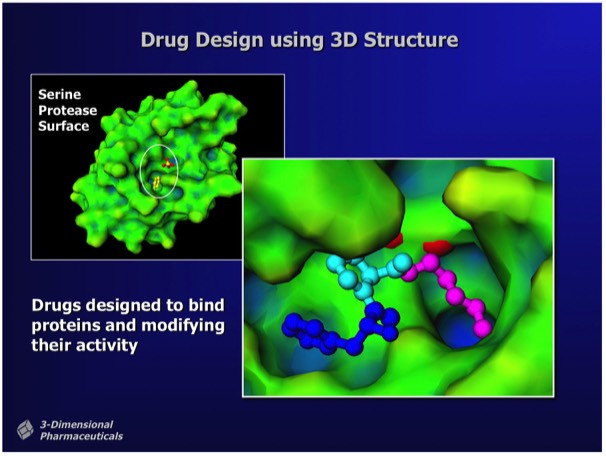
Structure-Based Drug Design uses the 3D structure of a drug lead bound to a target protein as a direct means to visualize drug-protein interactions and to determine chemical modifications that can improve potency. Although protein structures can in principle be used for ab initiio ligand design (e.g. using computational fragment docking methods), most leads are found using experimental methods like activity or binding screens.
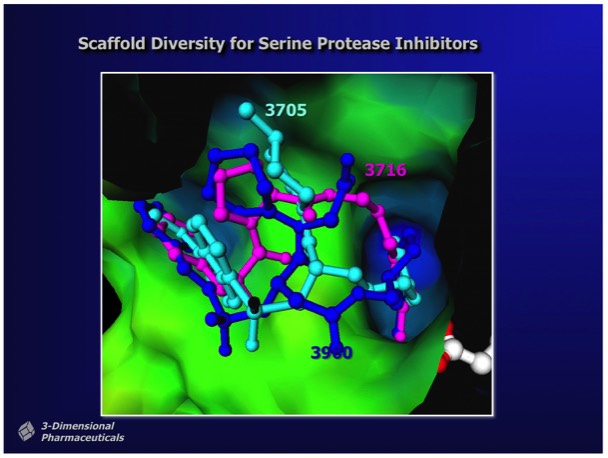
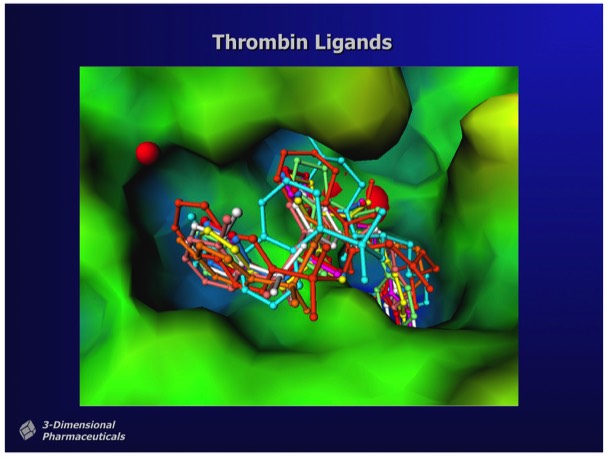
Structure-Based Design campaigns for drug binding optimization typically involve the determination of numerous protein-drug complexes. Although the design strategy usually focuses on optimizing specific interactions, X-ray results typically show that numerous small adjustments in both the ligand and active site structures occur even for small chemical changes in the binding ligand.
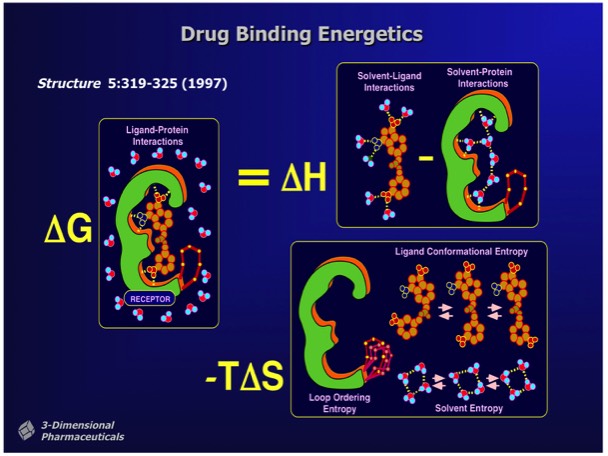
Changes in free energy that accompany ligand binding determine ligand affinity. Many factors can contribute to free energy changes that affect binding affinity. These include:
- Changes in bonding that occur between groups on the protein, the ligand and water.
- Changes in configurational entropy that occur in the protein, ligand, or water on ligand binding.
The major limitation of structure-based drug design is that many of the factors that contribute to ligand binding are difficult to quantitate based on static X-ray structures.
- Changes in bonding that occur between groups on the protein, the ligand and water.
- Changes in configurational entropy that occur in the protein, ligand, or water on ligand binding.
The major limitation of structure-based drug design is that many of the factors that contribute to ligand binding are difficult to quantitate based on static X-ray structures.
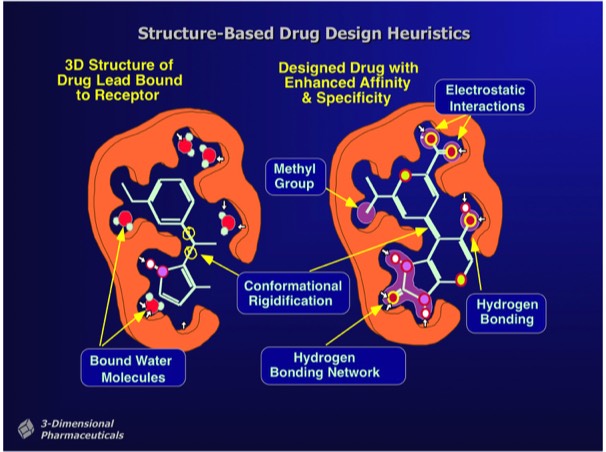
Observations of high-affinity protein ligand interactions create precedents for heuristic approaches to increasing ligand binding affinity. These often cue from observations of made on lead compounds bound in the protein binding site together with “bound” water molecules that can signal potential sites for ligand modification. Ligand modifications can introduce charge-charge interactions, hydrogen bonded interactions, or Van der Waals interactions depending on the environment of the bound water molecule or adjacent protein group. Some ultra-high affinity ligand interactions (e.g. streptavidin-biotin, some transition state analogs) involve charge redistributions in the ligand that allow the formation of extended hydrogen bond or dipole networks.
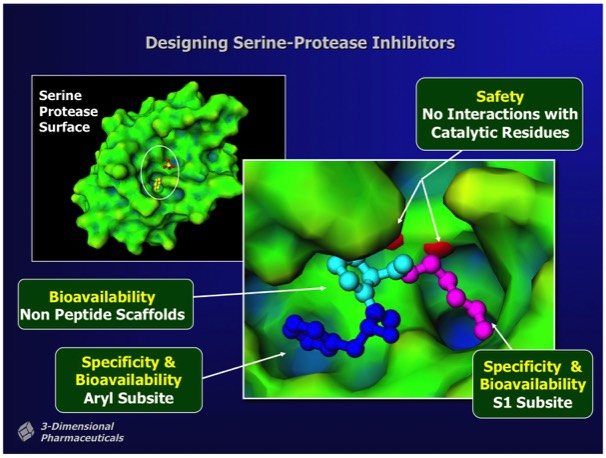
Structure-Based Design can also incorporate heuristic rules that lie outside the basic requirements of good binding energetics. For example, in designing inhibitors for thrombin, a serine protease involved in hemostasis that has subsite binding specificity for both basic and aromatic side chains, inhibitors can be designed that incorporate a non-peptidic linker to connect appropriate bio isosteres that bind at each subsite. Although enzyme transition state analogs make useful research tools, they generally have limited utility as drugs, owing to the high potential for off-target effects. This is a particular problem for thrombin transition state inhibitors owing to the high circulating concentration required to achieve adequate enzyme coverage in the circulation.
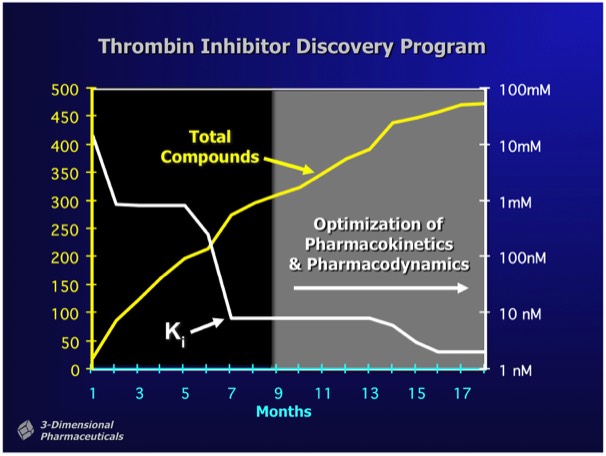
Depending on the nature of a target, initial HT screening hits may range in affinity from 500 nanomolar to 5 micromolar or more, although X-ray or NMR fragment screening can occasionally find ligands that bind with millimolar binding affinity. As shown as left, structure-based programs are effective at rapidly finding high-affinity compounds. Most importantly, SBDD allows the elaboration of lead compounds that preserve binding affinity but additionally possess characteristics required for preclinical development (e.g. bioavailability &ADMET).
Note that the majority of compounds synthesized are highly potent in vitro, but lack properties required for successful development.
Note that the majority of compounds synthesized are highly potent in vitro, but lack properties required for successful development.
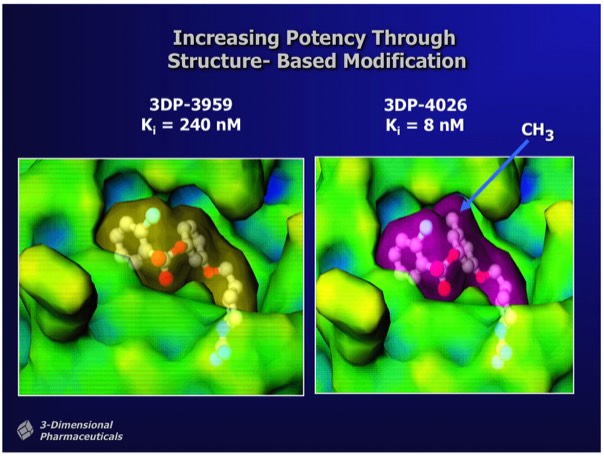
Significant changes in binding affinity can often accompany minor changes in drug structure. In the case of thrombin inhibitors shown, that differ by a single methyl group, the ~300x binding enhancement comes from a combination of improved hydrophobic interactions as well as conformational restriction of the methylated drug when bound in the enzyme active site. Experience has shown that one-by-one custom synthesis is the only reliable way to find compounds that inhibit enzymes with the single-digit nanomolar potency typically required for a compound that can be developed as a drug. Although efforts in recent years have conjectured advantages in screening libraries with more chemical "diversity", there is no evidence that this produces more frequent high-affinity hits or compounds that can be preclinically developed with fewer rounds of custom analog synthesis.
Most structure-based drug design programs are supported through computational evaluation and comparison of potential compounds for relative binding affinity. However, for reasons outlined above involved in computing binding free energy, quantitative evaluation of inhibitor binding behavior continues to be an elusive goal at the practical level. For many high-affinity interactions, such as the streptavidin-biotin interaction and binding of transition stste intermediates, ligand repolarization effects appear to be particularly important. Repolarization effects are absent from computations that employ point charge models most commonly used in computational chemistry calculations to guide structure-based docking or design programs.