Protein Substructure Motifs
The period from 1970 to 1980 saw a sharp increase in the number of protein structures determined and refined at high resolution using X-ray crystallography. The availability of multiple refined structures, generally accessible to the scientific community through the Protein Data Bank then run by Tom Koetzle at Brookhaven National Labs, made it possible to examine many structural features of proteins in increasing detail. This in turn enabled many key advances both in understanding protein structure and the development of new methods for protein modeling and structural refinement.
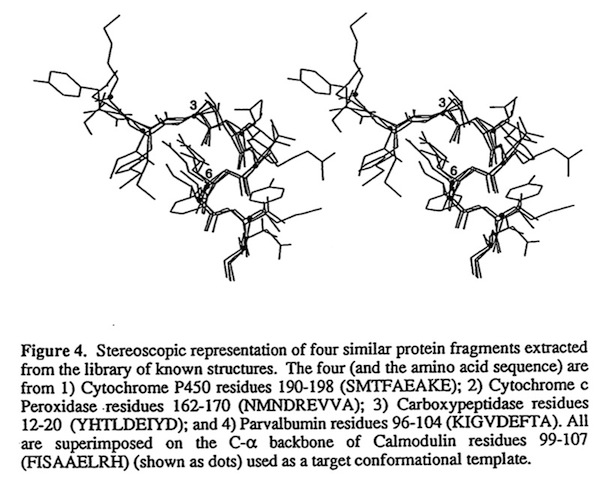
FRAGLE
Following emerging observations of recurrent structural motifs in proteins, I worked with Barry Finzel at DuPont CRD to develop one of the first fragment searching programs that allowed simultaneous application of c-alpha superposition and amino acid sequence correspondence to find local structural motifs and loops in refined protein structures. We used the approach both to improve the speed and accuracy of building structures into X-ray electron density maps and to define polypeptide conformations for modeling studies. The approach has subsequently become a key feature of protein modeling programs used for building structures into X-ray electron density maps, protein engineering, protein folding, and de novo design.
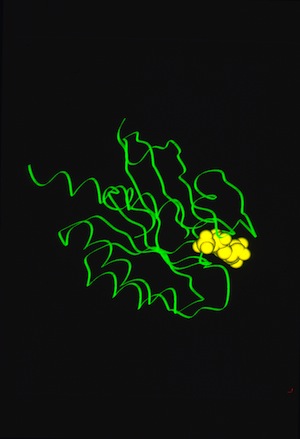

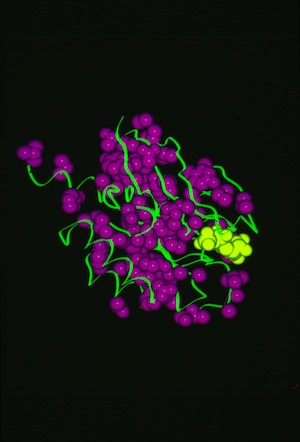
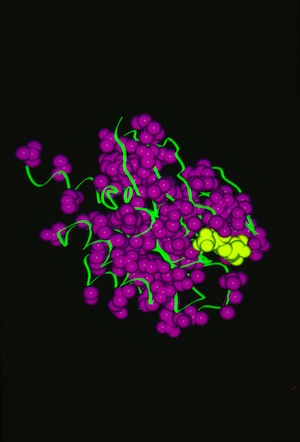
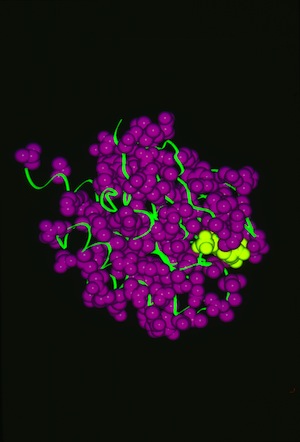
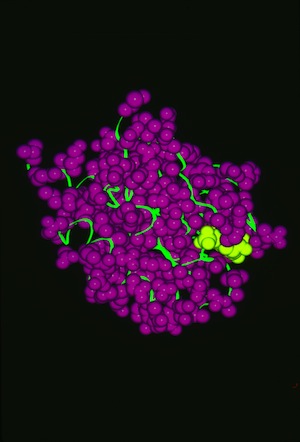
RAS P21 PROBIT Rebuilding
Structural data analyzed by Ponder and Richards in 1987 showed that protein-side chains tended to assume a limited set of canonical low energy rotamer conformations. Consequently John Wendoloski and I, noting that some amino acids were additionally restricted when present in specific secondary structure elements, developed a program PROBIT that used a secondary structure-dependent rotamer library to pack protein interiors from a backbone skeleton. The algorithm generated statistics for side-chain rotamer distributions depending on sequence and/or secondary structure context, and then used a probability-weighted approach to define which side chains were sequentially added to the backbone as the structure was rebuilt. The approach was quite effective in producing proteins with interior packing replicating the X-ray structure aspects of this approach has been incorporated into several subsequent protein modeling programs used for protein engineering or de novo protein design.