Protein Architecture
My introduction to structural biology began while working with Fred Richards and Hal Wyckoff at Yale University from 1962 to 1967. At that point in time relatively few protein structures were known, and very few known at high resolution. There were many basic questions whose answers we now know, but which were poorly understood at the time. My particular interests focused on understanding proteins essentially as . I was started on this course on reading Darcy Thomson’s book On Growth and Form, which illustrated how evolution had selected engineering solutions for problems in biological structure. For example, the illustration reproduced below shows the cross section of a condor wing bone, where material is deposited in the form of a Warren truss, a mechanical structure well known for its efficient use of material and resistance to bending moments.

Vulture Wing Bone in the form of a Warren Truss
As more structures were determined in the 1960’s and 1970’s, recurrent structural features were recognized in proteins. Many of these evidently occurred in the absence of significant amino acid sequence homology, suggesting that basic energetic or mechanical principles must be at work in protein structural organization. I became particularly interested long-range structural organization and trying to understand what the recurrence of structural motifs was trying to tell us about how proteins folded and maintained stable configurations with precise long-range structural organization. This interest lead to the examination of several classes of protein structural domains and recurrent structural features.
One observation of general interest was the situation of extended networks of contiguous H-bonding interactions, particularly in beta-sheet proteins. These were obvious features of tensile structures designed to resist deformation.
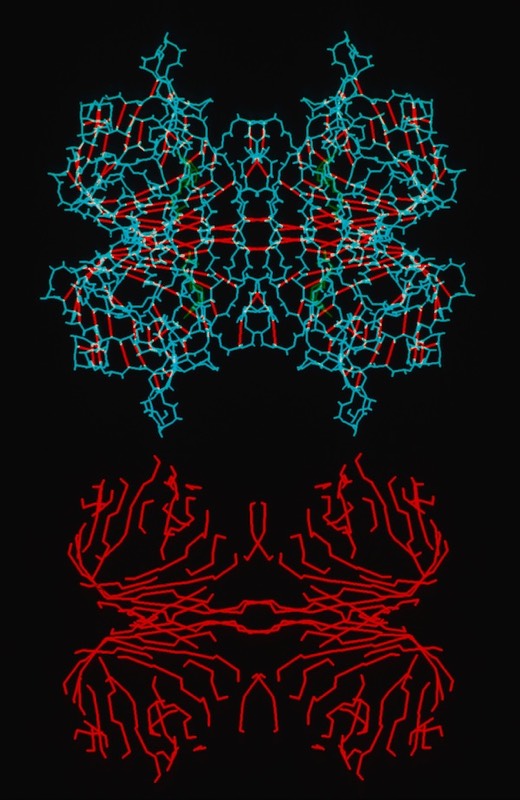
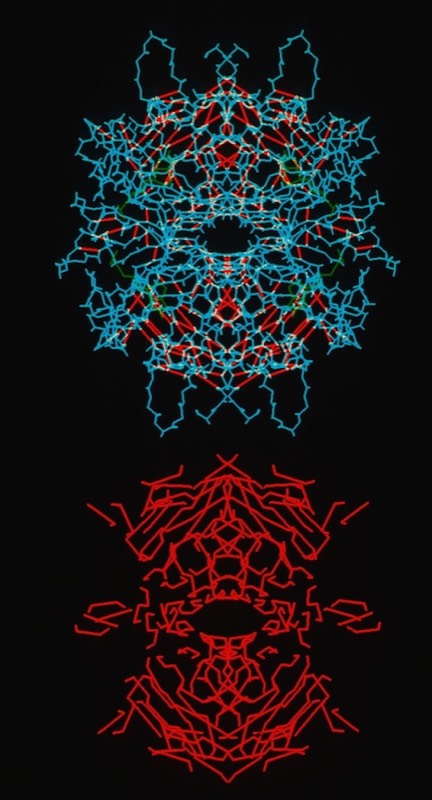
Streptavidin Tetramer Structure and Contiguous H-Bonding Lattice (Red)
Structural studies of proteins revealed that beta sheets in globular proteins seldom formed the flat structures inferred from structural studies of fibrous proteins like silk and keratin. My studies with Dave Weatherford defined the origins of the physical effects that produced the unusual variety of diverse beta-sheet geometries seen in globular proteins and produced atomic-scale models showing excellent correspondence with coordinates of beta sheets observed from X-ray crystallography. In addition, our studies demonstrated may aspects that were related to protein folding and dynamical behavior that were related to cooperative structural transitions in beta sheets.
The proliferation of X-ray structures determined in the decade of 1970 to 1980 revealed numerous recurrent structural motifs in proteins that were not obviously related to specific amino features of amino acid sequence. Instead, these were inferred to represent architectural features that were repeatedly selected by evolution for their structural stability or ability to fold efficiently. Following our structure determination of cytochrome c-prime from a photosynthetic bacterium, which folded in a 4-alpha helical motif, I worked with Pat Weber to develop and understanding of the structural properties of the 4-alpha helical bundle proteins, which show a great diversity of structural and functional diversity in nature.
As high resolution protein structures became more common in the 1980's, it became possible to examine local structural details in proteins, which revealed many recurrent local structural motifs, often associated with specific animo acid sequence signatures. I worked with Barry Finzel at DuPont CRD to develop one of the first fragment searching programs that allowed simultaneous application of c-alpha superposition and amino acid sequence correspondence to find local structural motifs and loops in protein structures. The approach has subsequently become a key feature of protein modeling programs used for building structures into X-ray electron density maps, protein engineering, protein folding, and de novo design.
Following the observation by Ponder and Richards in 1987 that protein-side chains tended to assume a limited set of canonical low energy rotamer conformations, and noting that some amino acids were additionally restricted when present in specific secondary structure elements, John Wendoloski and I developed a program PROBIT that used a secondary structure-dependent rotamer library to pack protein interiors from a backbone skeleton. The algorithm used a probability-weighted approach to define which side chains were sequentially added to the backbone as the structure was rebuilt. This approach has been incorporated into several subsequent protein modeling programs used for protein engineering or de novo protein design.